Identification and characterization of two consistent osteoarthritis subtypes by transcriptome and clinical data integration | March 2021 | Rheumatology (Oxford, England)
Rodrigo Coutinho de Almeida, Ahmed Mahfouz, Hailiang Mei, Evelyn Houtman, Wouter den Hollander, Jamie Soul, Eka Suchiman, Nico Lakenberg, Jennifer Meessen, Kasper Huetink, Rob G H H Nelissen, Yolande F M Ramos, Marcel Reinders, Ingrid Meulenbelt
Abstract
OBJECTIVE: To identify OA subtypes based on cartilage transcriptomic data in cartilage tissue and characterize their underlying pathophysiological processes and/or clinically relevant characteristics.
METHODS: This study includes n = 66 primary OA patients (41 knees and 25 hips), who underwent a joint replacement surgery, from which macroscopically unaffected (preserved, n = 56) and lesioned (n = 45) OA articular cartilage were collected [Research Arthritis and Articular Cartilage (RAAK) study]. Unsupervised hierarchical clustering analysis on preserved cartilage transcriptome followed by clinical data integration was performed. Protein-protein interaction (PPI) followed by pathway enrichment analysis were done for genes significant differentially expressed between subgroups with interactions in the PPI network.
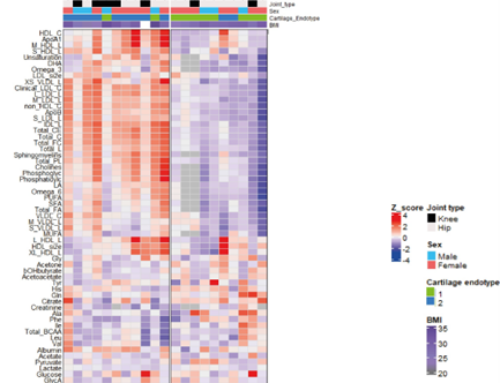
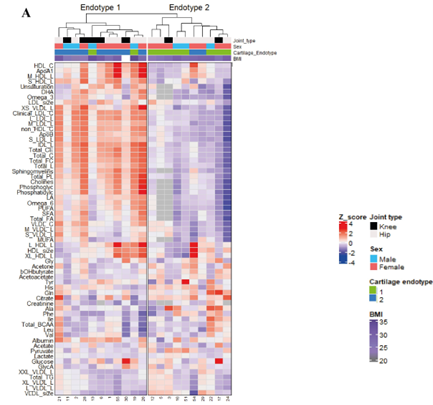
RESULTS: Analysis of preserved samples (n = 56) resulted in two OA subtypes with n = 41 (cluster A) and n = 15 (cluster B) patients. The transcriptomic profile of cluster B cartilage, relative to cluster A (DE-AB genes) showed among others a pronounced upregulation of multiple genes involved in chemokine pathways. Nevertheless, upon investigating the OA pathophysiology in cluster B patients as reflected by differentially expressed genes between preserved and lesioned OA cartilage (DE-OA-B genes), the chemokine genes were significantly downregulated with OA pathophysiology. Upon integrating radiographic OA data, we showed that the OA phenotype among cluster B patients, relative to cluster A, may be characterized by higher joint space narrowing (JSN) scores and low osteophyte (OP) scores.
CONCLUSION: Based on whole-transcriptome profiling, we identified two robust OA subtypes characterized by unique OA, pathophysiological processes in cartilage as well as a clinical phenotype. We advocate that further characterization, confirmation and clinical data integration is a prerequisite to allow for development of treatments towards personalized care with concurrently more effective treatment response.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Leave A Comment